Shingles
Shingles
Varicella-Zoster, Lifelong Companion
Sneaky, invasive,
Lurking serpent; the resident
Persistent menace.
Lots of different viruses can infect us. Mostly they are tiny little critters with a single strand of RNA, a few proteins, and a hard coat to hold it all together and allow it to stick to a cell and invade. We either inhale, ingest, or imbibe them into our body. They infect our cells and make millions of copies of themselves. Our immune system gets engaged to thwart them, and away they go, never to be encountered again. It’s one and done, hopefully without too dire a consequence.
But there is one group of viruses that do so much more. They can get into us in any number of ways besides the usual breathing and swallowing, and instead of going away when our immune system reacts to them, they literally become a part of us, establishing their double-stranded DNA in our cells’ nuclei. Some can remain in our body for our entire life. They are, of course, the herpes family of viruses, and the old saying “Herpes is forever” is right on the mark.
Compared to other viruses the herpes group is one of the most sophisticated. It’s not surprising, as the virus has been around for over a hundred million years. The nucleic acid is double-stranded DNA, just like ours, and there are over seventy proteins produced to protect, escort, and enable it. Most viruses undergo replication and multiplication in the cytoplasm, but the DNA of herpes viruses enters the host cell’s nucleus and operates from there. It is transcribed just like our DNA, using the host cell machinery to carry out its work.
The name herpes derives from the Greek word herpein, meaning “to creep.” The ancient Greeks used the word as a general description for maladies such as cold sores, lupus vulgaris, ringworm, eczema, and other skin conditions. The use of the word herpes as a more definitive diagnosis for the cutaneous disease we know today began in the early 1800s.
The first publication recognizing herpes simplex as a communicable disease was written by French dermatologist Jean Baptiste Emile Vidal in 1893. Despite the ubiquity and communicability of the disease, it wasn’t until the 1940s that the etiologic agent, a very large virus, was identified.
For most people the name herpes makes them think of a skin infection, either around the mouth or the genitalia. But in nature, there are over 100 known herpes viruses, nine of which infect humans with a range of infected sites. In the big picture, they are classified in the Family Herpesviridae. For humans, the herpes viruses encompass three groups, or Subfamilies, designated alpha, beta, and gamma. They are all similar in general structure and biology but differ in the diseases they cause.
The alpha herpes includes herpes 1 (mouth), herpes 2 (genitalia), and varicella-zoster, commonly called chickenpox and shingles. The beta Herpes includes the human pathogens cytomegalovirus (CMV), roseola virus, also known as herpesvirus 6A and 6B, and another roseola virus, herpesvirus 7. The gamma group includes the human Epstein Barr virus and Kaposi sarcoma-associated virus. Each group has its own characteristics, and the diseases they cause are unique to each strain.
Each human herpesvirus is assigned a number,
preceded by the letters HHV, for Human Herpes Virus:
HHV-1 Herpes Simplex 1
HHV-2 Herpes Simplex 2
HHV-3 Varicella Zoster
HHV-4 Epstein Barr
HHV-5 Cytomegalovirus
HHV-6A and B Roseola virus*
HHV-7 Roseola virus
HHV-8 Kaposi’s sarcoma-associated virus
*(6A and 6B have been determined to be distinct; hence 9 human Herpes viruses)
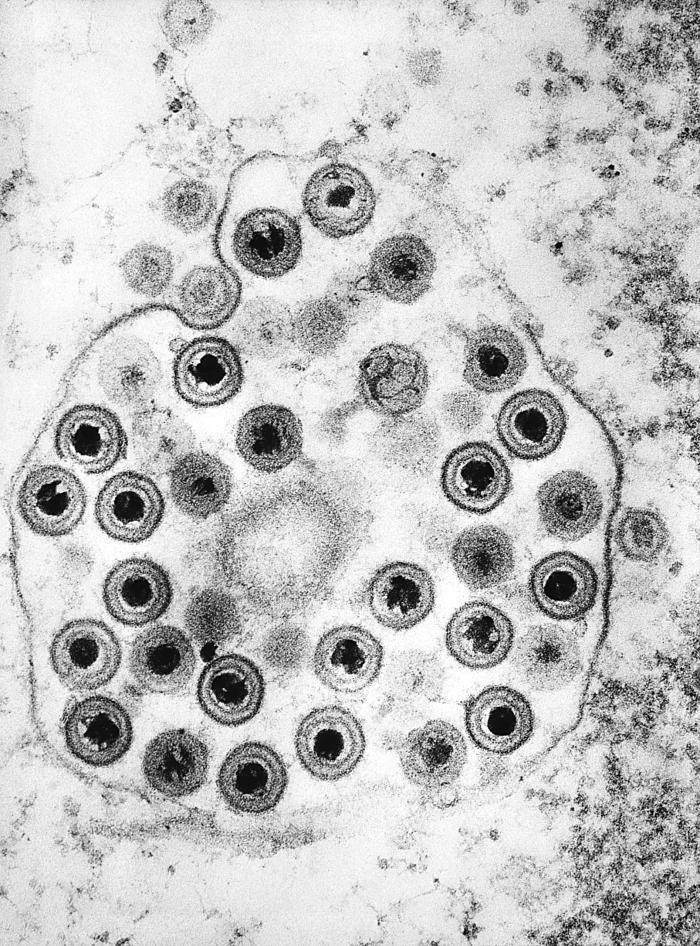
The members of the herpes family all have the same general structure. Inside is the DNA. It’s a fairly long, linear genome, capable of coding for 70 or more proteins. The genome is immediately surrounded and protected by a tough protein capsule. Made up of 6 different types of proteins, the capsule is very precise in its architecture, and it serves to surround, protect, and escort the DNA inside the host cell like a space capsule protects astronauts. Surrounding the payload of the DNA in its capsule is an amorphous layer known as the tegument. In the tegument are found about 20 proteins that carry out many of the virus’ jobs. On top of it all is a membrane not produced by the virus but obtained from the host cell's membrane as the virus escapes. Located in the membrane are around a dozen viral proteins, each associated with a carbohydrate, so they are given the name glycoproteins (“glyco” refers to the carbohydrate portion). They’re each designated by a lower-case g and upper-case letter designation, like gB and gD. These glycoproteins protruding through the outer membrane enable the virus to attach to specific receptors on the host cell. A few of them function in evading the immune system.
Herpes is a remarkable microbe, millions of years in the making. It infects in two distinct fashions, both beginning with the letter L. Lysis is a Greek word meaning to “loosen” or “break apart.” The lytic phase of a herpes infection refers to the virus’ ability to tear apart the host cell as the newly formed virions escape. Latent is from the Latin word latere, meaning to “lie hidden,” and that’s just what the virus does. In the latent phase the virus doesn’t produce the seventy-plus proteins needed for a new virus, but just a few to enable the viral DNA to reproduce in synchrony with the host cell and carry out a few other tasks. Since the nucleic acid is double-stranded DNA, just like ours, it rounds up and sits in the cell’s nucleus, right near the host cell chromosomes. It can remain there for decades, as anyone who has gone from chicken pox in childhood to shingles late in life can attest.
The first step in the cellular invasion of herpes is the virus's attachment to the host cell's membrane. The virus itself has a membrane that it got from the last cell invaded. Protruding through the surface of the viral membrane are about a dozen proteins, several of which work together to allow the fusion of the viral membrane to the host cell membrane, allowing for the penetration of the viral payload into the host cell.
The viral payload consists of the DNA packaged within its protective protein capsid, accompanied by the viral proteins of the tegument. Early in the host cell invasion stage, the viral DNA is linear, a long string with both ends unattached. The mission for the virus is to get this important bundle to the host cell nucleus. The integrity of the protective protein coat is vital, as DNA isn’t supposed to be in the host cell’s cytoplasm, only in the nucleus. Molecular sensors are ever-present in the host cell cytoplasm to detect DNA, and if encountered, chemical alarm bells go off, effectively aborting the virus’ pursuit. The protein coat prevents this.
Each tegument protein has an important job. For one, a couple of them hook the capsid onto a microtubule in the host cell. Just like a train on a track, the viral DNA package then makes its way up the microtubule directly to the host cell nucleus. A few other tegument proteins allow this to happen. Once it reaches the nucleus, the host cell's nuclear pore is opened to let the viral DNA in. Again, the accompanying tegument proteins are instrumental. As it enters the host cell nucleus, the capsid proteins are dispersed so the viral DNA enters by itself.
Once inside the host cell nucleus the linear viral DNA rounds up and forms itself into a circle, much like a plasmid in a bacterium. It’s considerably larger than a bacterial plasmid but not nearly as large as a human chromosome, so it is given the name episome. The viral DNA is transcribed into messenger RNA for translation into proteins, but not all at once. To do so would result in a sudden release of foreign protein inside the cell and the virus would come to a quick end. By a unique set of circumstances, transcription of viral DNA into RNA goes in stages, called immediate early, early, and late.
The first six proteins produced, those of the immediate early group, are instrumental in directing the activation of the next group of genes, known as the early genes. These earliest genes and their protein products direct the translation of the next group of genes, the so-called late genes, which produce proteins for viral protein production. The final stage is the reproduction of the viral genome. The whole process is carefully structured to allow for the proper amount of time between viral protein production. Making the late proteins too early would result in the misreading of the viral genome and alerting the host cell’s immune machinery.
While all this is happening, the virus has proteins that effectively shut down the host cell's metabolism. They are very efficient at it, and the host cell becomes a controlled virus factory. Once the many viral proteins are made and the DNA copied, the virions are assembled and make their way to the edge of the cell, to be released in a process that lyses the host cell. The preferred means of release by the virus is to enter a cell that abuts the original cell, thus avoiding the exterior and the immune defense. This is the lytic phase of the virus, as the host cell from which the virus is escaping breaks apart.
If the lytic phase of the family of herpes viruses were the whole story, we would probably consider them just something of a nuisance. Illness that lasts a few days and then goes away happens all the time, and often we don’t know what caused it. But herpes have perfected a unique feature that makes them a very challenging foe: latency. Being a double-stranded DNA virus that reproduces in the host cell nucleus allows them to remain with us for our whole lives. Usually, they just sit there doing nothing with our not being aware of their presence. But for some people, their long-term presence is potentially catastrophic.
Many of our cells die and are replaced all the time. They live out their usefulness and then are sloughed off or gobbled up by macrophages. It’s a normal process. For a virus to infect such a cell is a dead-end; it will die off with the cell. To be a lifelong inhabitant of a cell, the cell it dwells in must itself be lifelong. The herpes family has developed the ability to seek out such cells and infect them in addition to the short-lived cells where it undergoes lytic infection.
Nerve cells aren’t turned over. They last our lifetime, so it is no surprise that several herpes viruses can infect nerve cells. The alpha-herpes (simplex 1, simplex 2, and varicella) can all establish a latent infection in nerve cells. Similarly, beta- and gamma- herpes can infect a wide range of cells, inducing them to persist for the life of the infected individual.
The family of herpes viruses is very efficient in cell invasion and propagation. They have been infecting animals for over a hundred million years, and humans for as long as we have existed as a species, so they’ve had a long time to get it right. A “good” parasite doesn’t want to damage its host, at least not very much. The herpes viruses are classic examples of this. Well over half, maybe as many as 90% of humans, are infected by at least one member of the herpes family yet show no symptoms. The most apparent symptoms are displayed by the alpha-herpes group, with simplex 1 and 2 along with varicella displaying pathognomonic signs, but even these are usually short-lived in the initial lytic phase. That’s not to say the herpes viruses are entirely benign. Far from it. There is a very broad spectrum of the consequences of infection, from asymptomatic to fatal. The interaction of the virus and the immune system is critical in determining the course of the disease.
One of the first orders of business for the virus after it enters a new host cell is to shut down the cell’s protein synthesis. Several enzymes in the viral tegument disable the host cell’s RNA before it can be translated into protein at the ribosome. This has the effect of reducing the chances of apoptosis as well as curtailing cytokine production to alert the immune system. The enzymes that shut down host cell mRNA activity are among those produced late in the virus’ cycle, so viral mRNA is not impacted nearly as much as the host cell’s when the virus traverses the cytoplasm.
One of our most potent immune forces is the ability to display components of an invading virus on a cell’s surface. By doing this, T-cells and B-cells can be mustered to attack the virus when encountered. Normally, viral proteins are dismantled, and pieces of them, usually about 5-10 amino acids in length, are put onto an MHC molecule, which displays the peptide on its surface. The peptide-laden MHC molecule is then transported to the cell surface to openly express the little piece of virus. After several steps, the T-cells and B-cells that react with the displayed microbial peptides are activated, and the adaptive immune system is fully engaged to recognize and destroy the virus both inside and outside cells.
The herpes viruses have developed an elaborate system of proteins to greatly subvert this usually efficient system. The host cell has a series of proteins called TAP, which are responsible for attaching the viral peptide to the MHC molecule inside the cell in the endoplasmic reticulum. Herpes proteins interfere with the TAP process, and the MHC molecules never receive their cargo. Viral display on the infected cell's surface is abated, and the adaptive immune response is significantly curtailed.
Our cells can easily recognize double-stranded RNA because our cells do not make it and consider it a foreign substance. At some point in their life cycle inside the cell, RNA viruses create double-stranded RNA, so an immune response to RNA viruses is usually quite vigorous. But the herpes genome is double-stranded DNA, just like ours, so it doesn’t look like foreign material. Once the virus becomes latent, it shuts down protein synthesis, so there is no immune response to these foreign proteins. As long as the viral DNA can stay in the host cell nucleus it can remain in the latent state, going unnoticed by the immune system.
But DNA in the cytoplasm, outside the cell’s nucleus, is a sign of trouble. It means either an invading pathogen or a problem with the cell’s own metabolism. To detect and react to stray DNA in the cytoplasm cells, human cells have a most elaborate and complex device known as the inflammasome. Diagrams of inflammasomes look something like a spaceship from a science fiction movie, with a large round region on top and long dangling molecules protruding underneath. Inflammasomes don’t roam around the cell's cytoplasm, but subordinate components do. When one of them detects the presence of DNA, it becomes active, signaling other molecules that then, in a cascade fashion, form the inflammasome. After all this complicated business, the job of the inflammasome is twofold: to release the inflammatory cytokines interleukin-1 and interleukin-18, and the host cell is instructed to destroy itself along with the invading virus. The host cell’s auto-destruction is called pyroptosis. As the words “inflamed” and “pyro” suggest, cell destruction is not an orderly process like apoptosis. It is more traumatic, something of an explosive event. But the host cell dies, taking the invading microbe with it. The system is most used against bacteria since they contain DNA, but the herpes family is the viral exception.
Some herpes viruses have a means of subverting the formation and activity of inflammasomes. A herpes tegument protein, VP22, inhibits an integral part of the inflammasome, a substance called AIM2. Without AIM2, building the inflammasome and its important work is greatly curtailed.
The group of blood proteins known as complement is an important deterrent to invading microbes, including viruses. Complement can work alone against the intruder, or in concert with other members of the immune system such as antibodies and neutrophils. Complement is also vital in giving a chemical alarm about the presence and location of the invader. The family of herpes viruses have developed the means of reducing the effectiveness of complement on initial contact. One of the herpes virus’ surface glycoproteins can bind to the critical C3 and C5 complement components, blocking their activity. As a result, the body’s initial response to the invading virus is significantly reduced.
A very important group of immune cells working against intracellular pathogens like viruses are the natural killer lymphocytes (NK cells). By chemical sensors on their surface, NK cells can detect a serious problem inside an infected cell and set about the job of destroying it and its pernicious invader. By detecting the presence or absence of key molecules on the surface of cells, the NK lymphocytes can release their appropriate destructive molecular assassins and destroy the infected cell. For herpes viruses, this will not do. Over the eons they have developed the means of counteracting their nemesis NK lymphocytes. One important molecular signal of infected cells is known as MICA, which is displayed on the cell’s surface. It is an unequivocal signal to the NK lymphocyte that the cell displaying it needs to be killed. Some herpes viruses have developed molecules to ensure that MICA remains in the cell and the natural killer lymphocytes don’t encounter it and do their job. The virus, of course, survives. To add insult to injury, some herpes viruses induce the natural killer lymphocytes to kill themselves through apoptosis. The result is lots of virus remaining viable within the still-functioning host cell.
One of our most important cells for immunity are the dendritic macrophages, those starfish-looking cells that roam around seeking foreign invaders. They are very good at detecting pathogen-associated molecular patterns and internalizing the invading microbe. Once inside the DC cell, the microbe is systematically broken down and carried off to the nearest lymph node for introduction to the appropriate lymphocytes. Dendritic macrophages are especially good at picking up and removing microbes introduced into the skin, like many herpes viruses are. To survive, the viruses must overcome the action of the dendritic macrophages, and herpes is among the best at doing this. The virus can alter the maturation of the dendritic cell phagosome in which it resides, delaying the display of its protein parts on the phagocyte’s surface. The virus also interferes with the activity of the important MHC molecules, significantly limiting the recognition of the virus by the rest of the immune system.
All told the family of viruses known as the Herpesviridae are extremely efficient and communicable. Most of us have at least one of the nine members, and many have more than one. Just as there is a range of viruses in the family, there is also a wide range of symptoms and sequelae. Just how sick an individual becomes depends as much on the immune reaction to the virus as the invading virus itself.
Varicella-Zoster. One of the strangest names of a human disease is chickenpox. Strange in that it has nothing to do with chickens. They don’t get sick from the virus, harbor it, or become involved in any way. The most likely explanation for the name is that somewhere, sometime, someone thought the pox marks resembled the result of pecks by a chicken. Be that as it may, the disease has been known to just about every parent for centuries. It has been a very common childhood ailment and, thankfully, usually not too serious in its acute phase. Unfortunately, like other herpes viruses, it does not just go away after the initial illness. It remains with a person for the rest of their life, and for about 30% of us, it can emerge as something much more malevolent: shingles.
The virus has two official names, varicella and zoster, the former referring to the acute “chickenpox” stage, the latter to the shingles stage. In ancient times there was no way of knowing the two were caused by the same virus. In some ways, the childhood ailment resembles a very mild form of smallpox, or variola, hence the “vari” part of the name. The “cella” suffix means diminished or much reduced. Zoster is a Greek word for “belt” or “girdle,” since the disease shingles often, but not always, attacks the mid-abdomen. The virus is called varicella-zoster virus, usually abbreviated VZV.
Varicella has a typical herpes virus construction but with some unique characteristics. It is a little smaller than its alpha-herpes cousins simplex types 1 and 2, and it attacks by the respiratory route. The incubation period, the time from the first encounter with the organism to the first display of symptoms, is around 14 days, a long time for viruses. Most viral infections incubate just a few days. But just because no symptoms are displayed for two weeks after varicella enters the body doesn’t mean nothing is going on. Quite the contrary. The virus becomes very active almost immediately.
After entering the epithelial cells of the upper respiratory tract, it begins multiplying. The virus is best served by getting itself into lymph tissue, either a node or the tonsils. The cells that perform this task for it are the dendritic macrophages, which gobble up free virions and make their way to the lymph tissue. The dendritic cells are designed to digest the virus, display its entrails, and hook up with compatible lymphocytes, which can put an end to the menace. Instead, the dendritic cells serve as chauffeurs to carry the virus around the infected body unmolested. The virus goes from the initial epithelial cells it enters to dendritic macrophages, then to the nearest lymph tissue. It then enters either a lymphocyte or macrophage that carries it to every part of the body, including the macrophages of the reticuloendothelial system. It continues to replicate, making its way into the bloodstream, from whence it is deposited in the cells of the dermis of the skin.
This is quite a journey. From the respiratory epithelial cells to lymph tissue, to various bodily organs by way of the bloodstream, and back into the bloodstream to settle in the tissues of the skin and respiratory cells. All this is accomplished in the face of highly developed immune systems designed to halt such an incursion into the body. But varicella is highly successful in its quest because of its sophisticated armaments.
A major opponent of viral infections is apoptosis, programmed cell death. By killing itself, the cell, which is easily replaced, prevents the virus from using its internal machinery to make more copies of itself; the virus dies with the cell. So to thrive, varicella must be able to prevent apoptosis. On the other hand, after it has replicated and is ready to escape from the host cell, the virus needs to have the cell break apart through apoptosis to release it. Varicella has the capacity to do both in a remarkably sophisticated way. The virus has the proteins that promote the host cell’s apoptosis, but only after the virus has had time to replicate and make many copies of itself. This allows the easy release of the virus in an orderly fashion, thus preventing an inflammatory encounter. The virus can then be transported to its next destination. Just like many things in life, the timing of apoptosis is important, and varicella is a master of it.
Another important job of varicella is diminishing the amount of interferon and other cytokines produced by the host cell. Type one interferons signal cells throughout the body about the presence of a virus, and chemical processes are initiated to prevent the spread of the virus. Since varicella infects by going from cell to cell and in and out of the bloodstream, it must quell the amount of interferon produced. It is very good at doing this, with at least three proteins it manufactures designed to substantially halt or slow down both the production of and the response to interferon.
Interferon is powerful in its ability to help us contain invading viruses, but it has a downside: it makes us feel sick with body aches and fever. Interferon must be carefully controlled so that it is released in the proper amount only when necessary. A key protein in controlling the amount of interferon produced is known as suppressor of cytokine signaling 3, or SOCS3. The role of this cytoplasmic protein is to put a brake on excessive interferon production. Varicella has an enzyme that significantly increases the activity of interferon-suppressing SOCS3, thereby using the cell’s own chemical to mitigate interferon production.
Human cells infected by varicella should be sitting ducks for natural killer lymphocytes, the blood cell whose job is to destroy their fellow human cells infected by a virus. The signal to the natural killer lymphocytes that everything is all right within the cell it is surveying is the presence of the molecular complex MHC on the cell’s surface. Without enough MHC, the natural killer cell detects something is wrong and unleashes several killing molecules to liquidate the infected cell and its viral passenger. But varicella has devised ways to thwart the natural killer cells by modifying the ability of the NK cell to dock onto the surface of the damaged cell. Two stress-induced proteins, ULBP2 and ULBP3, are normally transported to the host cell’s surface, where they encounter the surveillance protein NKG2D on the natural killer cell. This enables the killer cell to release its deadly contents and destroy the virus-laden cell. But varicella has several proteins that incapacitate ULBP proteins, not allowing the NK cell to do its job. Instead of killing the cell containing the virus, natural killer lymphocytes can be used by the virus to transport it to other areas of the body. It’s kind of like a criminal hijacking a police car to take him to a crime scene.
With the influence varicella has over cell self-destruction and interferon production, it can reproduce and travel around our body with impunity. Ultimately it enters the cells of the dermis of the skin, replicates inside them, and then induces the cells to lyse. The result is what we see on the skin’s surface, pustules surrounded by a red ring. At first the fluid from the vesicle is clear, but after a short time it becomes thick and milky due to the presence of pus and fibrin. Viable virus is present in the fluid, and when the scabs break by itching or rubbing, the virus can become airborne and remain in the air for some time, allowing it to be transmitted to another individual. Upper respiratory tract cells are also infected, and the virus can be transmitted from there.
Like all herpes viruses, varicella can enter the latent state inside a host cell. The cell type it uses for this purpose is the neuron, which doesn’t undergo apoptosis. The virus has several proteins that ensure the infected neuron doesn’t destroy itself.
The peripheral nerves from our spinal column come in two major functions, motor and sensory. The motor nerves emanate from the front, or anterior, while the sensory from the back, or dorsal. It is these dorsal sensory nerve cells that varicella inhabits in the latent state. And, like their cousins herpes simplex type 1, they can dwell in the trigeminal nerve of the face. But unlike herpes simplex, which usually inhabits the lower two-thirds of the trigeminal, varicella is mainly found in the upper branch, which enervates the area around the eyes.
Just why varicella in its zoster form breaks out from its nerve cell host is not explained. It most likely has to do with an alteration in the immune status of the infected person, and several things can lead to that, the obvious ones being the aging process and heavy emotional stress.
Not everyone who had chickenpox will get shingles; only about 20-30% do. And not everyone is affected the same. Some cases are rather mild, more of an inconvenience, while others are severe, with a terrible rash, immense pain that doesn’t go away, and eye infections that can lead to blindness. Every case is different.
The area served by sensory nerves is called a dermatome. Rather than one specific point, a dermatome covers several centimeters of skin, extending in what looks like a band. It is within this dermatome that the zoster virus is expressed, first as localized pain and irritation, then as an ugly rash. The rash contains viable virus which can be communicated to other people. We don’t spread shingles, though; only chickenpox is spread to those who are susceptible.
Shingles near the eye can be devastating. Most times the infection occurs around the orbit of the eye, but sometimes the eye itself can be affected. Blindness sometimes results. Aggressive medical intervention with antiviral agents is vital.
Through the latter half of the 20th century, vaccines were developed for several infectious diseases usually associated with childhood: diphtheria, polio, pertussis, measles, mumps, and rubella can all be prevented by vaccination. Chickenpox was a prevalent childhood disease, but it was presumed by most that it was a mild disease, and a vaccination program wasn’t necessary. Since chickenpox is usually a more serious disease when contracted in adulthood, it wasn’t uncommon for parents to bring their children to a “chickenpox party,” where children would gather in a home afflicted by the disease in the hope that the child would get the disease and get it over with. The very name “chickenpox” suggests a trivial, unimportant malady.
That perception of chickenpox began to change throughout the 1960s and 70s as advances were made in chemotherapy. As steroidal and other chemotherapeutic medications were being used to treat previously untreatable diseases, it was becoming evident that varicella, once considered a benign virus, was, in fact, a killer. Children with leukemia could be treated with a drug that was successful, only to have the child die of varicella. Or a child could contract chickenpox, then visit a grandparent being treated with an agent that suppresses the immune system and unknowingly transmit a deadly disease to them. Vaccinating children for chickenpox would significantly reduce this threat.
Also in the 1960s and 70s, people were living longer. And the older we get, the more likely we are to contract shingles, and many more cases were being diagnosed and treated. Vaccines for varicella and zoster would also greatly improve public health.
In the 1960s professor Michiaiki Takahashi at Osaka University in Japan obtained specimens from a young boy who had chickenpox. The boy’s family name was Oka. In the laboratory, Dr. Takahashi placed the specimen containing the Oka varicella virus on tissue culture cells. Over the next several years the virus was passed to several different cell lines at sub-optimal temperatures. (Most cell cultures are maintained at body temperature, 37°C; Dr. Takahashi did his experiments at 34°C). Different cell lines were used, including human embryonic lung fibroblasts, guinea pig fibroblasts, and other cells from aborted human fetuses, to eventually isolate a strain of varicella that could infect, elicit an immune response, but not make the person sick. In other words, a live attenuated vaccine.
While it was not known then, the vaccine strain had at least eleven mutations in gene number 62, which codes for the viral glycoprotein gE. (The term open reading frame, or ORF, is often used for the term gene. In this case that would be ORF62). Without a properly functioning gE, the immune system easily contains the virus, and the typical chickenpox symptoms are not displayed, or only very slightly, although full immunity is established.
With the introduction of a live varicella virus questions immediately arose. Since it is a live virus, albeit attenuated, is it transmissible to others? Does the vaccine strain enter the latent stage, and if so, can it later re-emerge to cause zoster? If that were to happen, would it cause typical zoster or some abbreviated form? It had been long assumed that immunity to chickenpox was lifelong because people who had the disease as children would be continually exposed to the virus through contact with infected children, thus getting a natural booster of their immunity. If this were true, did the vaccine strain, if not transmissible, give this booster effect?
Because of the long timeframe of infection by varicella answers are not absolute, but it appears some 25 years after the introduction of the vaccine that it does indeed enter the latent stage and persists as the wild-type strain did. There is some evidence that the vaccine strain can emerge as a very mild form of shingles but not anywhere near the severity of the wild-type virus. It is speculation, but the persistence of humoral immunity is likely due to the periodic re-emergence of the virus from the latent stage, prompting an immune response but not yielding any clinical signs. If that is true, then continuous re-exposure to the virus in nature is not needed to keep up immunity. Whether a child immunized for chickenpox will need a vaccine for shingles when they are older is not known.
A September 3, 2024 article in the Wall Street Journal by health journalist Sumathi Reddy brings attention to the question of the effect of varicella vaccination versus natural infection and its effect on the development of shingles. Significantly more people under the age of 50 have recently been diagnosed with shingles. Beginning around 1998 the number of cases of shingles in people under 50 has increased, perhaps as much as a fourfold increase. The reason why remains to be elucidated. Factors such as the marked reduction of varicella in the population due to widespread vaccination, infection with COVID, and the greater use of immunosuppressive drugs have been proposed. Shingles is not a reportable disease to public health, so it may take some time to determine answers.
In the vast number of cases, chickenpox in children is a mild disease. In contrast, shingles in adults is very often a significant ailment. Approximately 30% of adults over the age of 50 develop shingles in their lifetime. Most cases are rather short-lived, a week or two duration, but the symptoms are considerably more troubling than chickenpox. A rash localized to the affected area and is not particularly problematic, just unsightly and with a small amount of itching. The more serious concern is the pain. The nerves affected are sensory, with pain receptors prominent. Having a mild or no rash with excruciating pain is not unusual. The duration of symptoms varies with the individual, with most over their infection in a couple of weeks. The good news for them is that usually shingles does not recur, and if it does, the second episode is not as severe as the first. The virus remains in the latent stage, but the cellular and humoral immunogenic burst protecting the patient is strong and persistent, preventing or minimizing future episodes.
Some people, though, have a life-altering situation when they develop shingles. When the virus emerges from the trigeminal nerve, it is usually from the upper branch, which enervates the eye region. Eye infections with varicella usually are in the area surrounding the eyes, the orbit. But in some cases, the eye itself is infected with the virus leading to severe medical problems that require a high level of care, with no guarantee of success. Another severe complication of shingles is a condition known as post-herpetic neuralgia, usually called PHN. The nerves in which varicella resides are sensory, which includes pain. Usually, the condition arises a little after the rash subsides, but it can occur without the rash. For some people, and it is impossible to predict just who, the pain is long-term and excruciating. Just a light touch is enough to send debilitating shock waves of pain. The duration of the symptoms is varied, sometimes for a few weeks, sometimes for life.
Just as we now have a vaccine for chickenpox in children, there is a vaccine for shingles in people over 50. (Vaccination is not currently recommended for people under 50 years of age, except for those with an immunologic indication). In fact, there are two shingles vaccines.
The first shingles vaccine manufactured is the same as the chickenpox vaccine, the live attenuated Oka strain. When given to children, the vaccine is designed to prevent chickenpox in children who have never had the disease. When given to older people, the vaccine is given to people who have presumably had chickenpox as a child and still harbor the latent form of the virus, leaving them potentially vulnerable to an outbreak of shingles. Their immunity to varicella has waned through the years, so giving them the chickenpox vaccine is meant to stimulate their immune response when/if the varicella virus reappears. The live shingles vaccine is given in a much higher dose than that given for chickenpox since the people it is administered to are much larger than children, and their immune response is not as great as that of a child. Recommended in 2006 for use in adults over 60, the live vaccine proved to be pretty good but certainly not perfect. Its effectiveness in preventing shingles was about 66%.
The main difference between the live vaccine strain of varicella and the wild-type virus is a marked alteration in the surface glycoprotein E. As the technology became available, it was possible to make altered glycoprotein E in the laboratory and administer just that one part of the virus instead of the whole thing. By itself, altered protein E is somewhat immunogenic, but the immune response advances significantly when an adjuvant is administered along with it. The adjuvant ASO1B contains two substances that substantially stimulate the immune response, both in cytokine production and the reaction of CD-4 lymphocytes, the greatest fighter against the cells containing varicella. Approved for use in the United States in 2017, the glycoprotein E plus adjuvant vaccine, known commercially as SHINGRIX, has replaced the live attenuated chickenpox vaccine for the prevention of shingles. While still early in its use, it has been shown to be over 90% effective in preventing shingles. One downside is that two doses are needed 2-6 months apart.
Varicella-zoster virus will always be with us. Vaccines will never eradicate it—they are not 100% effective, the virus is worldwide, and it is very easily spread. While not usually a cause for concern in healthy children, varicella can be devastating in children and older people who are immunocompromised, and shingles in some individuals is often a very serious, life-altering condition. It is a formidable foe.
The herpes viruses have double-stranded DNA, surrounded by a complex protein coat. Surrounding that is a tegument of around twenty viral proteins. A host cell-derived membrane with viral attachment proteins encases
the virus.

Contents
Introduction
Words to Remember
Sanitation Transformation
Florence Nightingale; Introduction of Sanitation
Louis Pasteur’s Game Changer
Pasteurization; Foundation of the Germ Theory
Nothing Small About It
Smallpox and the Beginning of Vaccination
Chemical Warfare
The History of Antibiotic Discovery
The Sad Saga of Mary Mallon
Typhoid’s Role in the Science of Epidemiology
What’s Bugging Us
General Description of Pathogenic Organisms
Little Sacs of Poison
Polymorphonuclear Neutrophils
First Responder
The Complement System
Safety Net
Macrophages and Dendritic Cells
Our Own Worst Enemy
Diseases Associated with Macrophages
The Attack of the Killer Lymphocytes
Description of Lymphocyte Activity
The Auxiliary
Acute Phase Proteins and Fever
The Big E
Escherichia coli, Shigella
GI Incursion
Salmonella and Typhoid Fever
A Most Difficult Infectious Disease
Clostridium difficile
Milk Bugs
Brucella and Listeria
Toxic Assault
Tetanus, Botulism, Diphtheria, Pertussis, Cholera
“The Scourge of Armies”
Typhus
Sucker Punch
Giardiasis
Freeloaders
Parasitic Diseases
The Kiss of Death
Trypanosomiasis
“Bad Air”
Malaria
Out of Africa
Ebola and Mosquito-borne Viruses
Reverse Assault
HIV
The World’s Greatest Unintended Consequence
Polio
Mind Control
Rabies and Toxoplasmosis
Kid’s Stuff
Measles
The Black Death
Bubonic Plague
Biological Weaponry
Anthrax
Under The Influence
Influenza
Lifelong Companion
Herpes Viruses
Pathogenic Shapeshifter
Coccidioidomycosis
Budding Trouble
Yeast Infections
Split Personality
Neisseria meningitidis
Crown of Thorns
Coronavirus
Pyogens
Streptococcus and Staphylococcus
Imperiled Infants
Respiratory Syncytial Virus, Rubella, Cronobacter
Peptic Predator
Helicobacter pylori
Consumption
Tuberculosis and Leprosy
Infection By Deception
Lyme Disease, Syphilis, Leptospirosis
Beating ‘em To The Punch
Vaccines
Hitting Them Where They Live
Antibiotics
Gut Check
Gut Microbiome
Afterword
References and further reading
The information in this blog was correct at the time of publication. The author does not assume any liability for loss or damage caused by errors or omissions.